Predictive Exploratory Synthesis
Exploratory synthesis in novel chemical spaces is the essence of solid-state chemistry. However, uncharted chemical spaces can be risky to explore experimentally, especially when materials synthesis is challenging. By coupling high-throughput computation and crystal structure prediction methods with large-scale materials informatics, we can survey, visualize, and explain stability relationships across broad compositional spaces—providing maps and chemical intuition to guide synthetic chemists in their quest to continuously extend the frontier of solid-state chemistry.
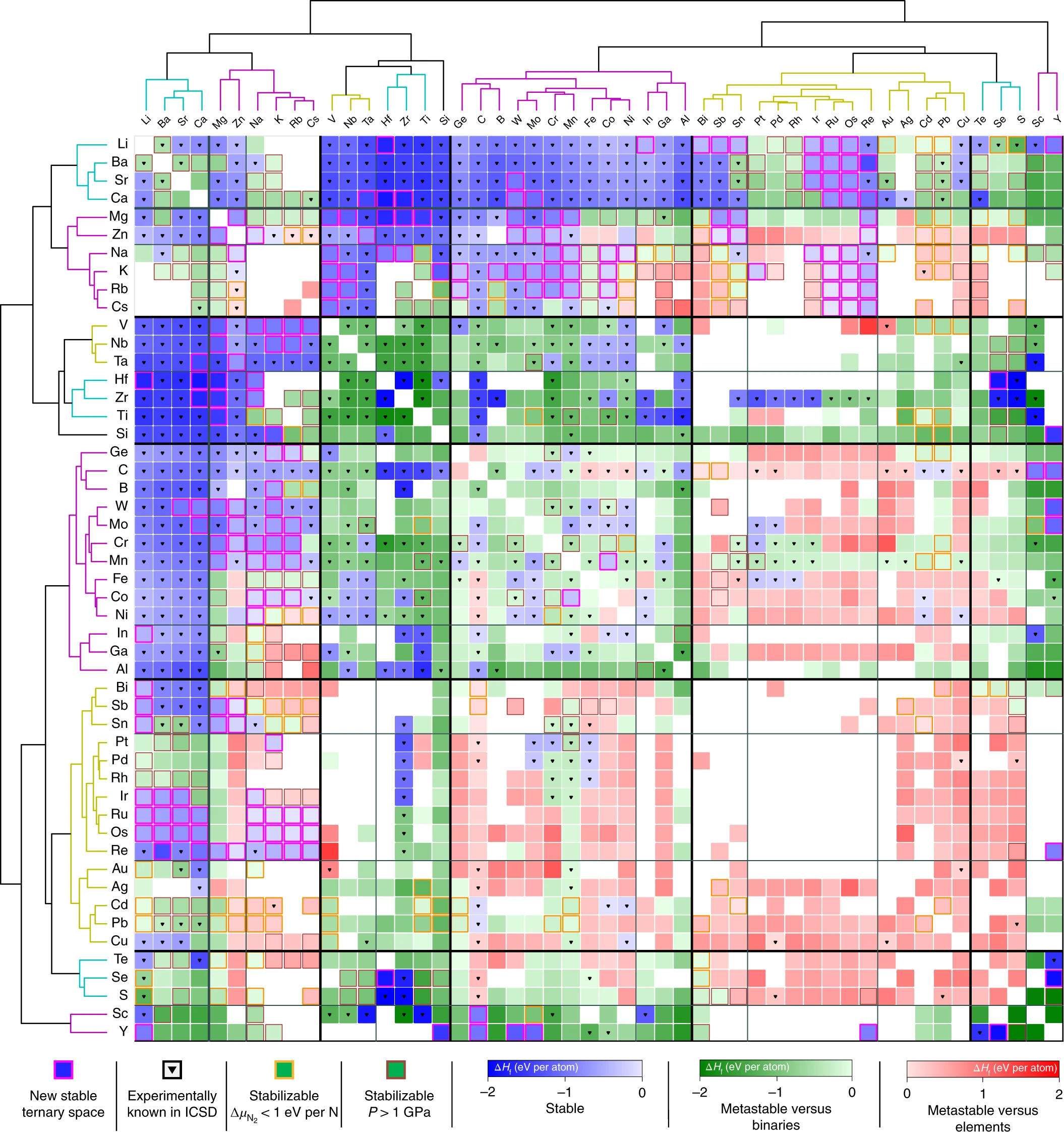
Recently, we constructed a large stability map of the ternary metal nitrides—an important class of functional materials that are rare in nature and difficult to synthesize in the laboratory. Using unsupervised machine-learning methods, we clustered the ternary metal nitrides into chemical families with distinct stability and metastability, (https://wenhaosun.github.io/TernaryNitridesMap.html), and predicted hundreds of new ternary spaces that are promising for further experimental investigation . Guided by the map, our experimental collaborators at the National Renewable Energy Laboratory synthesized 7 new Zn- and Mg-based ternary metal nitride thin-films. These novel earth-abundant nitrides are nondegenerate semiconductors, with exceptional optoelectronic properties which are tunable by a composition-modulated transition metal oxidation state. Our synthesized nitride films span a broad range of lattice parameters and band gaps in the visible regime, and hint at a new class of IIx-TM-Nitride materials—extending the III-N (Al/Ga/In) and II-IV-N (Zn-Ge/Sn) semiconductors.
Many of the machine-learning algorithms driving the AI revolution require billions of data points, whereas even large materials databases like the Materials Project only have 10^5 data points. With fewer data points, extracting physically meaningful insights from the ‘small-data’ space of materials requires more sophisticated material features than simple atomic descriptors like ionic radius, electronegativity, etc; or convenient DFT features like band gap or formation energy. Recently we developed new methods to extract from the DFT-computed electron density the mixed metallic, ionic, and covalent nature of solid-state bonding. These new and chemically-meaningful features allow us to interpret the electronic origins of thermodynamic stability and reactivity. Mining these electronic features can further yield fundamental new insights into structure-bonding-property relationships.
Select Publications
Sun, Wenhao, et al. "A map of the inorganic ternary metal nitrides." Nature materials 18.7 (2019): 732-739.
Sun, Wenhao, et al. "Thermodynamic routes to novel metastable nitrogen-rich nitrides." Chemistry of Materials 29.16 (2017): 6936-6946.
Aykol, Muratahan, et al. "Thermodynamic limit for synthesis of metastable inorganic materials." Science advances 4.4 (2018): eaaq0148.
Bauers, Sage R., et al. "Ternary nitride semiconductors in the rocksalt crystal structure." Proceedings of the National Academy of Sciences 116.30 (2019): 14829-14834.